 Nigel Sharfe 1,2† Harjit Dadi 1,2 Linda Vong 1,2 Jenny Garkaby 1 Laura Abrego Fuentes 1 Jessica Willett Pachul 1 Sandra Nelles 3 Amit Nahum 4,5 Chaim M. Roifman 1,2*
Nigel Sharfe 1,2† Harjit Dadi 1,2 Linda Vong 1,2 Jenny Garkaby 1 Laura Abrego Fuentes 1 Jessica Willett Pachul 1 Sandra Nelles 3 Amit Nahum 4,5 Chaim M. Roifman 1,2*Ori Scott 1† Nigel Sharfe 1,2† Harjit Dadi 1,2 Linda Vong 1,2 Jenny Garkaby 1 Laura Abrego Fuentes 1 Jessica Willett Pachul 1 Sandra Nelles 3 Amit Nahum 4,5 Chaim M. Roifman 1,2*
Background: STAT1 gain-of-function (GOF) is a primary immune dysregulatory disorder marked by wide infectious predisposition (most notably chronic mucocutaneous Candidiasis), autoimmunity, vascular disease and malignant predisposition. While atopic features have been described in some STAT1 GOF patients, they are not considered a predominant feature of the disease. Additionally, while eosinophilic gastrointestinal infiltration has been reported in some cases, this has always been described in the context of pre-existing oropharyngeal and/or esophageal Candidiasis.
Clinical cases: Herein, we report 3 members of a multi-generational family diagnosed with STAT1 GOF caused by a novel mutation in the N-terminal domain, c.194A>C (p.D65A). The proband presented initially with a long-standing history of treatment-refractory eosinophilic esophagitis (EoE) without preceding gastrointestinal tract fungal infections, and her mother was diagnosed with esophagitis as well.
Conclusion: EoE has been previously associated with alterations to STAT6 and STAT3 signaling pathways. The current report expands the possible association between JAK/STAT-related disorders and EoE, suggesting that EoE could be a primary disease manifestation of STAT1 GOF, even in the absence of oropharyngeal and/or esophageal Candidiasis.
Signal Transducer and Activator of Transcription (STAT) is a family of 7 structurally homologous transcription factors, with diverse functions both within and outside of the immune system. The canonical activation of STAT proteins follows a common sequence whereby extracellular receptor ligation prompts recruitment and cross-phosphorylation of tyrosine kinases of the Janus Kinase (JAK) family. This, in turn, creates a docking site for STAT proteins which are phosphorylated by JAK and undergo oligomerization to form active transcriptional complexes (1, 2). Within the STAT family, STAT1 plays a pivotal role in the signaling of various cytokines, most notably interferons and IL-27 (1–4). Over the past 15 years, monogenic disorders in the STAT1 gene have been described including biallelic loss of function (LOF) causing a profound combined immunodeficiency (CID) (5–7), a dominant negative LOF resulting in Mendelian Susceptibility to Mycobacterial Disease (MSMD) (8, 9), and an autosomal dominant gain-of-function (GOF) (10, 11). To date, over 400 cases of STAT1 GOF have been reported, making it the most common genetic cause identified in patients with chronic mucocutaneous Candidiasis (CMCC) (12, 13).
Since the initial description of STAT1 GOF as a syndrome of CMCC and hypothyroidism, its phenotypic spectrum has expanded dramatically to involve CID, intracranial vascular disease, various autoimmune manifestations, and malignant predisposition (10–17). Atopic manifestations have also been reported in STAT1 GOF patients, with a recent systematic review estimating 13.6% of patients to be affected by such features (13). Atopic presentations reported to date have included eczema, asthma and food allergies (12, 15, 18–21). Despite the above, atopy and eosinophilic disorders are still not commonly considered a predominant feature of STAT1 GOF, as opposed to other disorders affecting JAK/STAT signaling such as STAT3 LOF, STAT5B LOF or GOF, and JAK1 GOF (22). While gastrointestinal eosinophilia has been reported in a few STAT1 GOF patients (15, 23), this was typically identified following diagnosis of oropharyngeal/esophageal Candidiasis, and may have represented a secondary tissue reaction to fungal infection. Herein, we report a family of 3 generations affected by STAT1 GOF, with eosinophilic esophagitis (EoE) in the proband, and with esophagitis diagnosed in her mother as well.
Data compiled prospectively and retrospectively from medical records were entered into the Canadian Centre for Primary Immunodeficiency Registry and Tissue Bank, which has been approved by the SickKids Research Ethics Board (protocol no. 1000005598). All patients provided written informed consent.
Lymphocyte proliferative responses to mitogens, including PHA, anti-CD3 and anti-CD28 antibodies, were determined by thymidine incorporation, as reported previously (24). All assays were performed in triplicate and were compared with random normal controls.
Genomic DNA was isolated from patient peripheral blood leukocytes using the Geneaid genomic DNA extraction kit (Geneaid Mini Kit; Sensi Capital Corp, Toronto, Ontario, Canada). Patient mutations were diagnosed via a clinical panel (Inborn Errors of Immunity Panel, Prevention Genetics), and confirmed by STAT1 Sanger sequencing using DTCS Quick Kit on an automated sequencer (Beckman-Coulter CEQ 8000).
STAT1-deficient U3A cells were obtained from ATCC. pCMV6-STAT1 was from OriGene. The STAT1 D65A mutant was created using QuikChange II XL site-directed mutagenesis (Agilent) and confirmed by sequencing. U3A cells were transfected with cDNA for wild type STAT1 or D65A STAT1 using Lipofectamine 3000 (ThermoFisher Scientific), according to the manufacturer’s instructions. After 24 hours, transfected cells were serum-starved for 3 hours prior to stimulation with 100 ng/mL IFN-γ or 4ng/ml IFN-α. Whole-cell lysates were prepared in RIPA buffer, and 10ug per sample were loaded and analyzed by immunoblotting. Anti-phospho-Stat1 (Tyr701; 7649), anti-Stat1 (9172) and anti-GAPDH (2118) were all purchased from Cell Signaling Technology (MA, USA). Immunoblotting experiments were done in triplicates. All blots derive from the same experiment and were processed in parallel.
U3A cells transfected with wild type or D65A STAT1 were stimulated for 8 hours with either IFN-γ or IFN-α as described above, and mRNA levels of CXCL10 and CXCL9 were determined by quantitative real-time PCR. Expression data were normalized to the levels of the house-keeping gene GAPDH and are presented as mean and standard deviation from a total of 3 experiments. Statistical analysis was performed using the unpaired Student’s t-test.
A 39-year-old female presented to our clinic for further work-up in the context of prolonged and treatment-refractory EoE. The patient first developed symptoms of dysphagia to solids in her late adolescence, but was not formally evaluated until the age of 31 when the dysphagia progressed to involve both solids and liquids, and following multiple episodes of food bolus impaction. Two initial upper endoscopies revealed a macroscopically abnormal esophagus with multiple strictures and rings (Figure 1); however, no eosinophilia was noted at the time, and no organisms were identified. Testing for H. pylori was negative. Special stains for acid-fast bacilli and fungal elements were negative. As the patient continued to suffer from symptoms highly suggestive of EoE requiring esophageal dilations every few months, she underwent a repeat endoscopy at the age of 34, this time demonstrating eosinophil-rich esophagitis (20-22 eosinophils per high-power field in both proximal and distal esophagus), with eosinophil clustering and degranulation, and again no organisms noted and negative fungal stain. She was formally diagnosed with EoE, which has been difficult to treat over the next 5 years. The patient was initially trialed on a six-food elimination diet, with no substantial symptomatic relief. She was subsequently treated with a regimen of inhaled fluticasone, montelukast, and a proton pump inhibitor (PPI), without success. The patient was finally switched to a regimen of budesonide slurry and a PPI, resulting in improvement in her esophageal eosinophilia, although she continues to require frequent dilations. Notably, during this period she developed one episode of Candida esophagitis, which was treated with Nystatin and has not recurred. This was her first and only life-time episode of esophageal Candidiasis, with no oropharyngeal thrush ever reported.
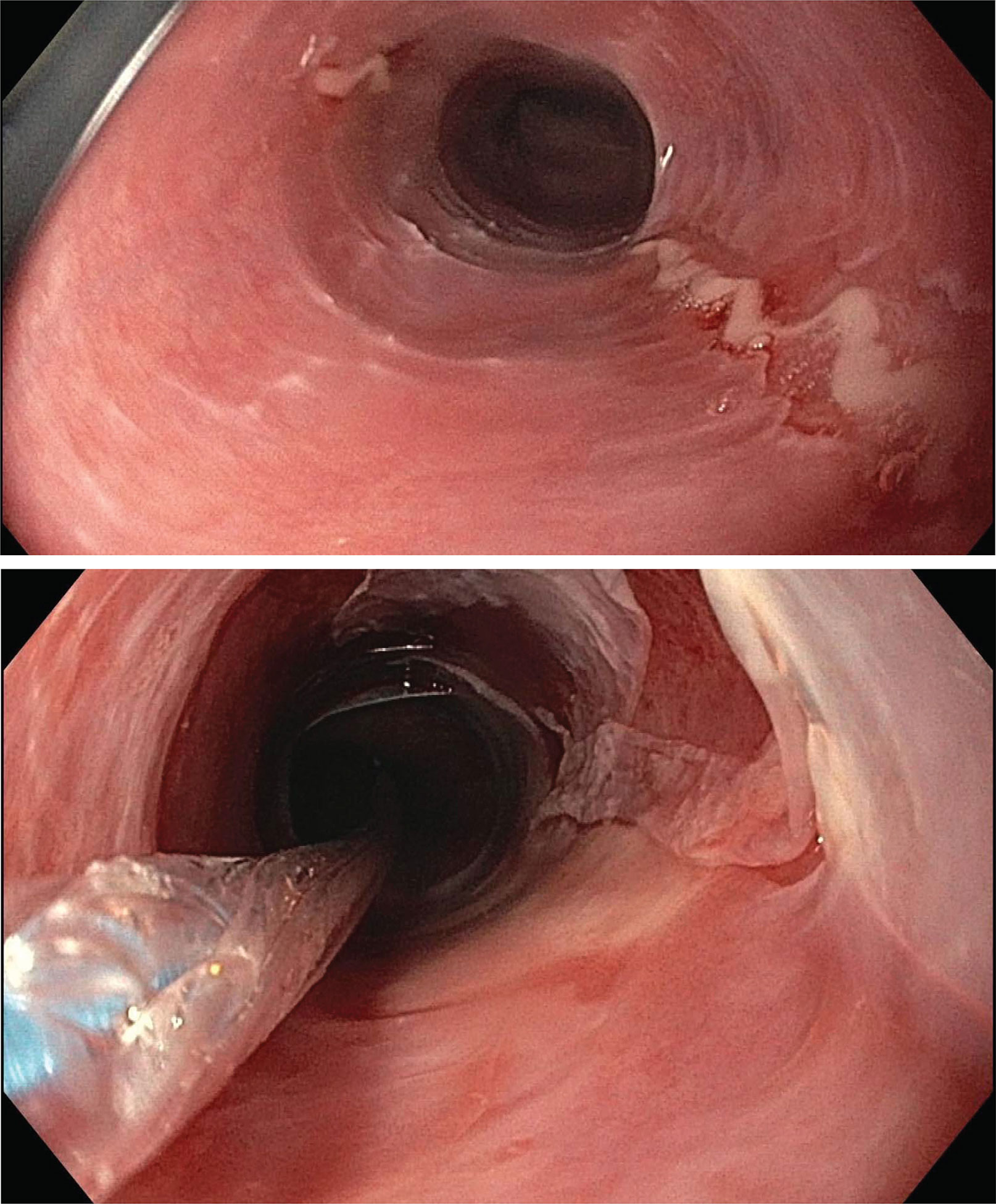
Figure 1 Endoscopic assessment of esophagitis in proband. Upper endoscopic assessment of the esophagus in our proband given history of recurrent esophageal strictures. Top: a ringed esophagus with linear ulcerations. Bottom: post balloon dilation, demonstrating sloughed, paper-thin esophageal mucosa without any evidence of deeper tears.
On our review of the patient’s infectious history, she endorsed a long-standing history of vaginal Candidiasis in her adult life, occurring several times per year. The infections typically responded to over-the-counter treatment with miconazole or clotrimazole, but recurred shortly after treatment. She also reported on previous fungal scalp infections, requiring the use of a medicated coal tar/chloroxylenol-based shampoo. She denied any other fungal infections, and had no history of viral, bacterial, Mycobacterial or opportunistic infections. She had no autoimmune, endocrine, autoinflammatory or malignant disease, and no other atopic features. She had received all age-appropriate vaccinations without difficulty. Review of the patient’s family history revealed that her mother (further described below) had a history of CMCC, and had recently been diagnosed with chronic esophagitis requiring dilations. The patient’s father had type 2 diabetes mellitus, hypertension, and coronary artery disease. Parents were both Canadian of mixed English-Irish ancestry, and non-consanguineous. The patient’s 3 half-siblings (1 maternal half-brother and 2 paternal half-siblings) were all healthy. One of the patient’s daughters (5 years old; further described below) was noted to have CMCC, recurrent acute otitis media (AOM) and atopic dermatitis. The patient’s three other children (ages 4-17 years) were healthy. A family pedigree is outlined in Figure 2A.
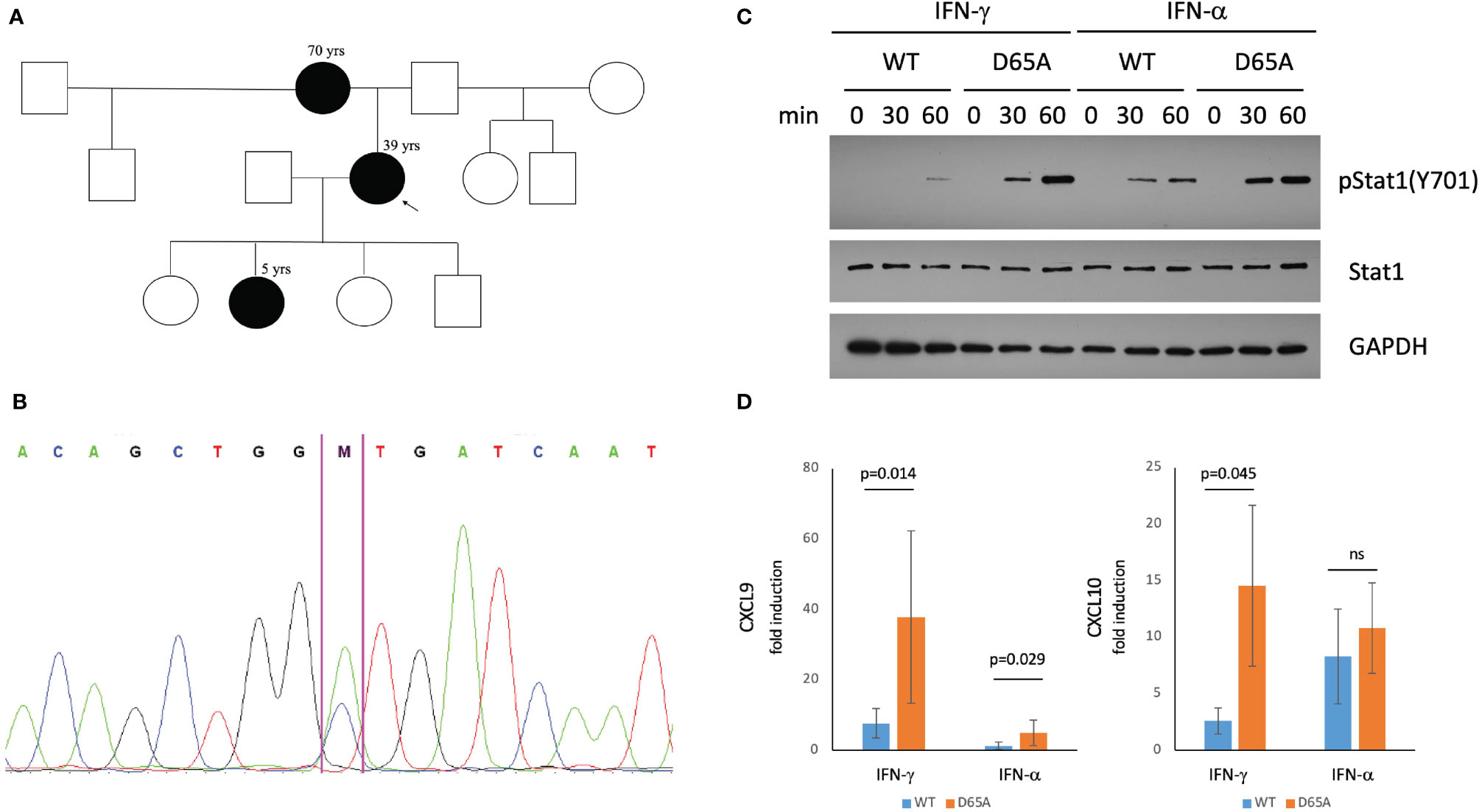
Figure 2 A STAT1 mutation identified in our patients causes STAT1 gain-of-function. (A) Pedigree of a three-generational family affected by STAT1 mutation (black). The proband (arrow) suffered from eosinophilic esophagitis and chronic mucocutaneous Candidiasis (CMCC). Her mother had a history of life-long esophagitis and CMCC. The proband’s daughter was affected by CMCC, recurrent acute otitis media, and atopic dermatitis. (B) Confirmatory sequencing of the STAT1 gene in our patients revealed a heterozygous mutation, c.194A>C (p.D65A) in the N-terminal domain. (C) Immunoblotting of STAT1-null U3A cells transfected with either wildtype or D65A STAT1 demonstrated normal total levels of STAT1 in the mutant, with elevated levels of pSTAT1 (Tyr701) following stimulation with either IFN-γ or IFN-α. (D) Quantitative real-time PCR demonstrated increased fold induction of CXCL9 in D65A U3A cells after stimulation with either IFN-γ or IFN-α, with increased fold induction of CXCL10 following stimulation with IFN-γ. ns, not significant.
An immune evaluation of the proband (detailed in Table 1) was normal, including complete blood count and differential (CBC-Diff), lymphocyte subset analysis, total immunoglobulins, vaccine-specific antibody titres, and T-cell mitogen stimulation responses.
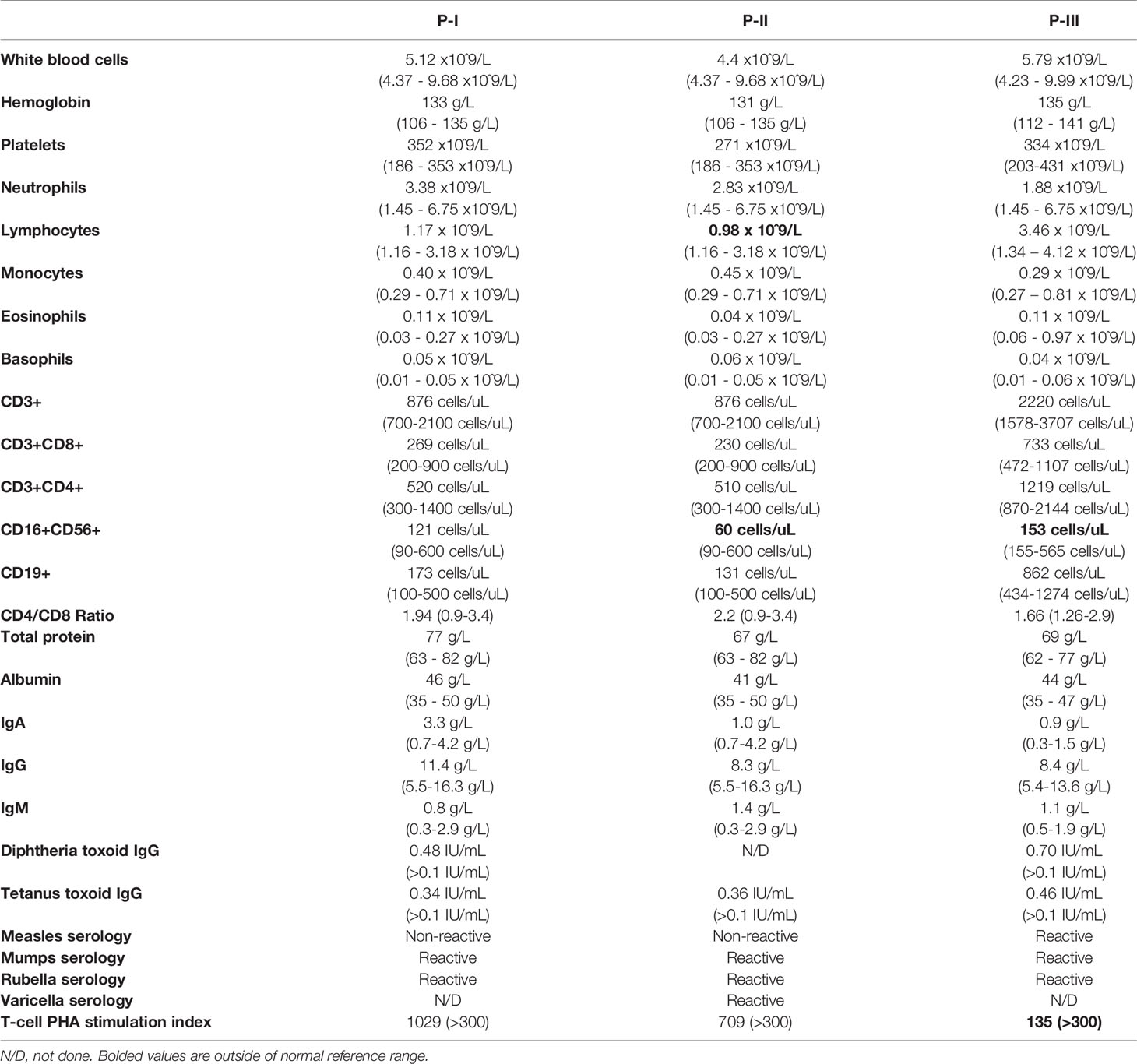
Table 1 Immune laboratory values for patients.
This 70-year-old woman reported on a 50-year history of dysphagia and choking episodes, but was only evaluated by a gastroenterologist for the first time at the age of 66. Endoscopic assessment showed a severely narrowed esophagus, with biopsy demonstrating extensive tissue fibrosis, rare eosinophils in the proximal esophagus, and no organisms identified. At that time, there was evidence of marked basal cell hyperplasia, which 3 years later progressed to a squamous cell papilloma, currently managed conservatively. On review of infectious history, the patient had suffered from CMCC since the age of 5 years. In the first decade of life this included episodes of oral thrush, whereas in adulthood she suffered from recurrent skin fungal infections, with no oropharyngeal, vaginal or nail infections noted. She never had any autoimmune or endocrine concerns. Laboratory evaluation in this patient was notable for lymphopenia, with reduced natural killer (NK) cell counts. Her evaluated B and T-cell responses were within normal range (Table 1).
This 5-year-old girl was affected by CMCC since early infancy, including persistent oral thrush in the first year of life, followed by recurrent fungal scalp infections throughout early childhood. She also had a history of recurrent AOM infections with subsequent hearing loss necessitating tympanostomy tube insertion, but no other sinopulmonary infections. This patient had no concerns for dysphagia or feeding difficulty. There was no history of autoimmunity or endocrinopathy. Her immune evaluation showed a borderline low NK cell count, as well as decreased T-cell proliferation in response to PHA stimulation (Table 1).
All patients underwent a genetic evaluation via a clinical Inborn Errors of Immunity panel (PreventionGenetics), demonstrating a novel heterozygous missense variant in the N-terminal domain of STAT1: c.194A>C (p.D65A; Figure 2B). This variant has not been reported in large population databases or literature. Another missense mutation affecting the same amino acid residue, c.193G>A (p.D65N) was recently reported as causing STAT1 GOF (25).
Immunoblotting assessment of U3A cells transfected with either wildtype or D65A STAT1 revealed comparable levels of total STAT1 between wildtype and D65A. While no pSTAT1 was seen at baseline following serum-starvation, stimulation with either IFN-γ or IFN-α resulted in a substantial increase in pSTAT1 in the mutant compared with wildtype, for both timepoints measures (30 and 60 minutes; Figure 2C), confirming a GOF phenotype. In regards to gene expression, we evaluated the induction of two known STAT1 targets, CXCL9 and CXCL10, following stimulation with either IFN-γ or IFN-α. The fold induction of CXCL9 was significantly higher in D65A compared with wildtype cells, after either IFN-γ or IFN-α (p=0.014 and p=0.029, respectively). Fold induction of CXCL10 was significantly increased in D65A after stimulation with IFN-γ (p=0.045) but not IFN-α (Figure 2D).
Following their diagnosis of esophagitis, both the proband and her mother have continued to require frequent esophageal dilations, as response to other standard EoE therapies including swallowed steroids, PPI and elimination diet has been limited. The possibility of initiating treatment with a JAK inhibitor has been considered by our team; however, there has been no clear indication for initiation of such a therapy in this family. Specifically, no severe autoimmunity, lymphoproliferation or severe refractory infections have been reported by the family, whereas atopy or EoE have not been well-studied to date as indications for treatment with JAK inhibitors. The proband’s mother is also followed conservatively for esophageal squamous cell papilloma, as described above. The proband’s daughter currently requires no treatment.
This report presented a family affected by STAT1 GOF with severe, treatment-refractory EoE as the presenting manifestation in the proband. Esophagitis requiring multiple dilations was also diagnosed in the proband’s mother, while the proband’s daughter displayed eczema, but had not yet developed esophagitis at the age of 5.
The association between EoE and STAT proteins, in particular STAT6, was first described in 2003 given the role of STAT6 in IL-4 and IL-13 signaling, and the demonstration that STAT6 deficient mice were resistant to an experimental model of IL-13-induced EoE (26). A decade later, functional studies in esophageal-derived cell cultures from EoE patients showed that STAT6 was required for transcriptional activation of the eosinophil chemoattractant, eotaxin 3 (27), an important player in EoE pathophysiology (28). Additionally, STAT6 inhibition using JAK inhibitors such as Ruxolitinib or Leflunomide resulted in significant reduction in eotaxin 3 expression (29). From a genetic standpoint, a genome-wide association study identified a significant association of EoE with variants at the STAT6 locus (30). Moreover, a recent study of pediatric EoE patients revealed that patients harbouring certain STAT6 variants were at a higher risk of disease relapse after a year of PPI treatment, compared with children not carrying such variants (31).
While variants in STAT6 have been identified in otherwise healthy individuals with EoE, a connection between primary immunodeficiency disorders (PIDD) and EoE has also emerged in recent years. Reports of EoE in patients with common variable immunodeficiency (CVID) were initially published in 2016 (32), with one population database study showing a higher prevalence of both CVID and IgA deficiency among patients with EoE (33). In 2018, a survey of the United States Immunodeficiency Network (USIDNET) identified 63 patients with eosinophilic gastrointestinal disease (EGID), of whom 50 had EoE. Almost half of the patients identified had CVID, while others had diagnoses of autosomal dominant hyper IgE syndrome (AD-HIES), chronic granulomatous disease, or CID. However, approximately one third of the patients had oropharyngeal/esophageal Candidiasis, and it was unknown what proportion of those had developed EoE prior to the onset of EGID (34). Within the realm of STAT-related PIDD, a National Institute of Health (NIH) study evaluating gastrointestinal manifestations in patients with AD-HIES identified high rates of esophageal eosinophilic infiltration, with other abnormalities including food impaction, esophageal tortuosity, ulcerations, or strictures requiring dilations, suggestive of possible EoE in this patient population (35).
Given the above-described association between AD STAT3 LOF and esophageal disease/EoE, it stands to reason that STAT1 GOF might cause EoE given loss of STAT3 promoter binding activity in this disorder. Additionally, STAT1 itself may also be implicated in EoE, given its role in IL-5 signaling in eosinophils (36, 37). Work by Nguyen et al. examined esophageal and peripheral blood samples of untreated EoE patients, comparing them with treated EoE patients as well as healthy controls. They found that transcript levels of not only STAT6 but also STAT1 were elevated in esophageal tissue of untreated EoE patients compared with control. Additionally, peripheral eosinophils and T-cells both showed enhanced phosphorylation of both STAT1 and STAT6. The authors suggested that this may be in keeping with the current understanding of EoE as a “mixed-type” immunological disorder, involving both Th2 and non-Th2 components (38).
From a therapeutic standpoint, JAK-inhibitors have emerged in recent years as a non-curative option for STAT1 GOF patients, and a potential bridge to HSCT, which may alleviate some immune dysregulatory features (20, 39, 40). However, data on their efficacy with respect to atopic disease manifestations in STAT1 GOF is scarce. Furthermore, concerns have been raised regarding increased predisposition to opportunistic and invasive infections as a result of treatment with JAK-inhibitors (41). Therefore, further evidence may be required prior to initiating treatment with JAK-inhibitors for EoE in the context of STAT1 GOF.
In summary, the current report describes a novel mutation in the STAT1 N-terminal domain leading to a GOF phenomenon, and with a concurrent predominant presentation of eosinophilic esophagitis in the proband, as well as esophagitis in her mother. The main limitations of the current report are its single-family scope, and the possibility that EoE and STAT1 GOF are concurrently present in a non-causative manner. Indeed, it is conceivable that the proband and her mother both developed esophagitis which is STAT1-independent. Further reports identifying this association, as well as rigorous mechanistic studies, would be required to establish with certainty whether and how STAT1 GOF may cause EoE. We suggest maintaining a high index of suspicion for EoE in STAT1 GOF patients presenting with dysphagia or food impaction, even in the absence of oropharyngeal/esophageal Candidiasis. We further propose that STAT1 GOF be considered in the work-up of patients presenting with refractory or atypical EoE, especially when esophagitis is familial, and in the context of other features of STAT1 GOF such as CMCC.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by SickKids research ethics board (protocol 1000005598). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Patient clinical evaluation by OS, JG, LA, JW, SN, AN, and CR. Laboratory analysis performed by NS and HD. The first manuscript draft was written by OS. All authors critically revised and approved of the final version of the manuscript.
This work was supported by Immunodeficiency Canada, The Canadian Centre for Primary Immunodeficiency and The Jeffrey Modell Research foundation (CMR). Salary support for OS has been provided by the Ontario Ministry of Health Clinician Investigator Program, the Hospital for Sick Children Clinician Scientist Training Program, the Canadian Child Health Clinician Scientist Training Program, and the Canadian Institutes of Health Research (CIHR) Doctoral Award: Frederick Banting and Charles Best Canada Graduate Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and Consequences of Jak–STAT Signaling in the Immune System. Nat Immunol (2017) 18(4):374. doi: 10.1038/ni.3691
2. Stark GR, Darnell JE Jr. The JAK-STAT Pathway at Twenty. Immunity (2012) 36(4):503–14. doi: 10.1016/j.immuni.2012.03.013
3. Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons From Interferons for Cytokine Responses. Front Immunol (2017) 8:29. doi: 10.3389/fimmu.2017.00029
4. Michalska A, Blaszczyk K, Wesoly J, Bluyssen HA. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front Immunol (2018) 9:1135. doi: 10.3389/fimmu.2018.01135
5. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired Response to Interferon-Alpha/Beta and Lethal Viral Disease in Human STAT1 Deficiency. Nat Genet (2003) 33:388–91. doi: 10.1038/ng1097
6. Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, et al. Human Complete Stat-1 Deficiency is Associated With Defective Type I and II IFN Responses In Vitro But Immunity to Some Low Virulence Viruses In Vivo. J Immunol (2006) 176:5078–508. doi: 10.4049/jimmunol.176.8.5078
7. Vairo D, Tassone L, Tabellini G, Tamassia N, Gasperini S, Bazzoni F, et al. Severe Impairment of IFN-Gamma and IFN-Alpha Responses in Cells of a Patient With a Novel STAT1 Splicing Mutation. Blood (2011) 118:1806–17. doi: 10.1182/blood-2011-01-330571
8. Kong XF, Ciancanelli M, Al-Hajjar S, Alsina L, Zumwalt T, Bustamante J, et al. A Novel Form of Human STAT1 Deficiency Impairing Early But Not Late Responses to Interferons. Blood (2010) 116:5895–906. doi: 10.1182/blood-2010-04-280586
9. Kristensen IA, Veirum JE, Moller BK, Christiansen M. Novel STAT1 Alleles in a Patient With Impaired Resistance to Mycobacteria. J Clin Immunol (2011) 31:265–71. doi: 10.1007/s10875-010-9480-8
10. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-Of-Function Human STAT1 Mutations Impair IL-17 Immunity and Underlie Chronic Mucocutaneous Candidiasis. J Exp Med (2011) 208(8):1635–48. doi: 10.1084/jem.20110958
11. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 Mutations in Autosomal Dominant Chronic Mucocutaneous Candidiasis. N Engl J Med (2011) 365(1):54–61. doi: 10.1056/NEJMoa1100102
12. Toubiana J, Okada S, Hiller J, Oleastro M, Gomez ML, Becerra JC, et al. Heterozygous STAT1 Gain-of-Function Mutations Underlie an Unexpectedly Broad Clinical Phenotype. Blood (2016) 127(25):3154–64. doi: 10.1182/blood-2015-11-679902
13. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, et al. Clinical Relevance of Gain-And Loss-Of-Function Germline Mutations in STAT1: A Systematic Review. Front Immunol (2021) 12:655. doi: 10.3389/fimmu.2021.654406
14. Sharfe N, Nahum A, Newell A, Dadi H, Ngan B, Pereira SL, et al. Fatal Combined Immunodeficiency Associated With Heterozygous Mutation in STAT1. J Allergy Clin Immunol (2014) 133(3):807–17. doi: 10.1016/j.jaci.2013.09.032
15. Uzel G, Sampaio P, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant Gain-of-Function STAT1 Mutations in FOXP3 Wild-Type Immune Dysregulation–Polyendocrinopathy–Enteropathy–X-Linked–Like Syndrome. J Allergy Clin Immunol (2013) 131(6):1611–23. doi: 10.1016/j.jaci.2012.11.054
16. Breuer O, Daum H, Cohen-Cymberknoh M, Unger S, Shoseyov D, Stepensky P, et al. Autosomal Dominant Gain of Function STAT1 Mutation and Severe Bronchiectasis. Respir Med (2017) 126:39–45. doi: 10.1016/j.rmed.2017.03.018
17. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The Extended Clinical Phenotype of 26 Patients With Chronic Mucocutaneous Candidiasis Due to Gain-of-Function Mutations in STAT1. J Clin Immunol (2016) 36(1):73–84. doi: 10.1007/s10875-015-0214-9
18. Faitelson Y, Bates A, Shroff M, Grunebaum E, Roifman CM, Naqvi A. A Mutation in the STAT1 DNA-Binding Domain Associated With Hemophagocytic Lymphohistocytosis. LymphoSign J (2014) 1(2):87–95. doi: 10.14785/lpsn-2014-0004
19. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic Stem Cell Transplantation in Patients With Gain-of-Function Signal Transducer and Activator of Transcription 1 Mutations. J Allergy Clin Immunol (2018) 141(2):704–17. doi: 10.1016/j.jaci.2017.03.049
20. Vargas-Hernández A, Mace EM, Zimmerman O, Zerbe CS, Freeman AF, Rosenzweig S, et al. Ruxolitinib Partially Reverses Functional Natural Killer Cell Deficiency in Patients With Signal Transducer and Activator of Transcription 1 (STAT1) Gain-of-Function Mutations. J Allergy Clin Immunol (2018) 141(6):2142–55. doi: 10.1016/j.jaci.2017.08.040
21. Erdős M, Jakobicz E, Soltész B, Tóth B, Bata-Csörgő Z, Maródi L. Recurrent, Severe Aphthous Stomatitis and Mucosal Ulcers as Primary Manifestations of a Novel Stat1 Gain-of-Function Mutation. Front Immunol (2020) 11:967. doi: 10.3389/fimmu.2020.00967
22. Lyons JJ, Milner JD. The Clinical and Mechanistic Intersection of Primary Atopic Disorders and Inborn Errors of Growth and Metabolism. Immunol Rev (2019) 287(1):135–44. doi: 10.1111/imr.12727
23. Eren Akarcan S, Ulusoy Severcan E, Edeer Karaca N, Isik E, Aksu G, Migaud M, et al. Gain-Of-Function Mutations in STAT1: A Recently Defined Cause for Chronic Mucocutaneous Candidiasis Disease Mimicking Combined Immunodeficiencies. Case Rep Immunol (2017) 2017:1–7. doi: 10.1155/2017/2846928
24. Arpaia E, Shahar M, Dadi H, Cohen A, Roifman CM. Defective T Cell Receptor Signaling and CD8+ Thymic Selection in Humans Lacking Zap-70 Kinase. Cell (1994) 76(5):947–58. doi: 10.1016/0092-8674(94)90368-9
25. Chen X, Xu Q, Li X, Wang L, Yang LU, Chen Z, et al. Molecular and Phenotypic Characterization of Nine Patients With STAT1 GOF Mutations in China. J Clin Immunol (2020) 40(1):82–95. doi: 10.1007/s10875-019-00688-3
26. Mishra A, Rothenberg ME. Intratracheal IL-13 Induces Eosinophilic Esophagitis by an IL-5, Eotaxin-1, and STAT6-Dependent Mechanism. Gastroenterology (2003) 125(5):1419–27. doi: 10.1016/j.gastro.2003.07.007
27. Zhang X, Cheng E, Huo X, Yu C, Zhang Q, Pham TH, et al. Omeprazole Blocks STAT6 Binding to the Eotaxin-3 Promoter in Eosinophilic Esophagitis Cells. PloS One (2012) 7(11):e50037. doi: 10.1371/journal.pone.0050037
28. Hogan SP, Mishra A, Brandt EB, Foster PS, Rothenberg ME. A Critical Role for Eotaxin in Experimental Oral Antigen-Induced Eosinophilic Gastrointestinal Allergy. Proc Natl Acad Sci (2000) 97(12):6681–6. doi: 10.1073/pnas.97.12.6681
29. Cheng E, Zhang X, Wilson KS, Wang DH, Park JY, Huo X, et al. JAK-STAT6 Pathway Inhibitors Block Eotaxin-3 Secretion by Epithelial Cells and Fibroblasts From Esophageal Eosinophilia Patients: Promising Agents to Improve Inflammation and Prevent Fibrosis in EoE. PloS One (2016) 11(6):e0157376. doi: 10.1371/journal.pone.0157376
30. Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS Identifies Four Novel Eosinophilic Esophagitis Loci. Nat Commun (2014) 5(1):1–5. doi: 10.1038/ncomms6593
31. Mougey EB, Nguyen V, Gutiérrez-Junquera C, Fernández-Fernández S, Cilleruelo ML, Rayo A, et al. STAT6 Variants Associate With Relapse of Eosinophilic Esophagitis in Patients Receiving Long-Term Proton Pump Inhibitor Therapy. Clin Gastroenterol Hepatol (2021) 19(10):2046–53. doi: 10.1016/j.cgh.2020.08.020
32. Chen M, Ko HM, Riffle ME, Andreae DA, Cunningham-Rundles C, Chehade M, et al. Eosinophilic Esophagitis Diagnosed in a Patient With Common Variable Immunodeficiency. J Allergy Clin Immunol Pract (2016) 4(5):995–7. doi: 10.1016/j.jaip.2016.03.023
33. Peterson K, Firszt R, Fang J, Wong J, Smith KR, Brady KA. Risk of Autoimmunity in EoE and Families: A Population-Based Cohort Study. Off J Am Coll Gastroenterol ACG (2016) 111(7):926–32. doi: 10.1038/ajg.2016.185
34. Ruffner MA, Garabedian EK, Fuleihan RL, Sullivan KE, Spergel JM. Eosinophilic Gastrointestinal Disease in Patients With Primary Immunodeficiency. J Allergy Clin Immunol (2018) 141(2):AB24. doi: 10.1016/j.jaci.2017.12.077
35. Arora M, Bagi P, Strongin A, Heimall J, Zhao X, Lawrence MG, et al. Gastrointestinal Manifestations of STAT3-Deficient Hyper-IgE Syndrome. J Clin Immunol (2017) 37(7):695–700. doi: 10.1007/s10875-017-0429-z
36. Pazdrak K, Justement L, Alam R. Mechanism of Inhibition of Eosinophil Activation by Transforming Growth Factor-Beta. Inhibition of Lyn, MAP, Jak2 Kinases and STAT1 Nuclear Factor. J Immunol (1995) 155(9):4454–8.
37. van der Bruggen T, Caldenhoven E, Kanters D, Coffer P, Raaijmakers JA, Lammers JW, et al. Interleukin-5 Signaling in Human Eosinophils Involves JAK2 Tyrosine Kinase and Stat1 Alpha. Blood (1995) 85(6):1442–8. doi: 10.1182/blood.V85.6.1442.bloodjournal8561442
38. Nguyen T, Gernez Y, Fuentebella J, Patel A, Tirouvanziam R, Reshamwala N, et al. Immunophenotyping of Peripheral Eosinophils Demonstrates Activation in Eosinophilic Esophagitis. J Pediatr Gastroenterol Nutr (2011) 53(1):40. doi: 10.1097/MPG.0b013e318212647a
39. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the Treatment of Immune Dysregulation in Patients With Gain-of-Function Signal Transducer and Activator of Transcription 1 (STAT1) or STAT3 Mutations. J Allergy Clin Immunol (2018) 142(5):1665–9. doi: 10.1016/j.jaci.2018.07.020
40. Bloomfield M, Kanderová V, Paračková Z, Vrabcová P, Svatoň M, Froňková E, et al. Utility of Ruxolitinib in a Child With Chronic Mucocutaneous Candidiasis Caused by a Novel STAT1 Gain-of-Function Mutation. J Clin Immunol (2018) 38(5):589–601. doi: 10.1007/s10875-018-0519-6
41. Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-Associated Infections: A Systematic Review and Meta-Analysis. Am J Hematol (2018) 93(3):339–47. doi: 10.1002/ajh.24976
Keywords: STAT1, heterozygous mutation, eosinophilic esophagitis, candidiasis, atopy, immune dysregulation
Citation: Scott O, Sharfe N, Dadi H, Vong L, Garkaby J, Abrego Fuentes L, Willett Pachul J, Nelles S, Nahum A and Roifman CM (2022) Case Report: Eosinophilic Esophagitis in a Patient With a Novel STAT1 Gain-of-Function Pathogenic Variant. Front. Immunol. 13:801832. doi: 10.3389/fimmu.2022.801832
Received: 25 October 2021; Accepted: 05 January 2022;
Published: 20 January 2022.
Megan Anne Cooper, Washington University in St. Louis, United States
Lisa Renee Forbes, Baylor College of Medicine, United States
Ofer Zimmerman, Barnes-Jewish Hospital, United States
Copyright © 2022 Scott, Sharfe, Dadi, Vong, Garkaby, Abrego Fuentes, Willett Pachul, Nelles, Nahum and Roifman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
† These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.